The Drug That Stops Cancer Also Teaches It How to Escape
ISB researchers reveal a darker side of targeted therapy: the same oncogene inhibition that shuts down cancer growth program can also ignite a stress-driven identity switch — revealing an early escape route that may shape the future of cancer treatment
Targeted therapy is often described as a battle against mutations. A drug hits its target, tumors shrink, and the cells that survive are the ones that happened to carry the right genetic changes.
But that is only part of the story.
New research from ISB and collaborators shows that some cancer cells don’t wait for mutations to rescue them. Instead, they adapt — rapidly and reversibly — by changing who they are. And strikingly, the same targeted therapy that shuts down cancer oncogenic drivers also helps create the stress conditions that launch this escape program.
In a study published in Nature Communications, ISB researchers and collaborators reveal how melanoma cells exposed to targeted therapy can slip into a temporary, drug-tolerant state within days of treatment. Rather than drifting randomly toward resistance, the cells follow a coordinated program — one that unfolds in a precise sequence and leaves behind a lasting molecular imprint.
A ‘Molecular Movie’ of Cancer Adaptation
To understand how this process begins, the research team turned to an approach that is still relatively rare in cancer biology: watching the system evolve in real time.
Using dense, time-resolved multi-omics measurements combined with computational modeling, the team reconstructed what they describe as a “molecular movie” of melanoma cells responding to therapy. Instead of comparing snapshots before treatment and after resistance emerges, they captured the transition as it happened.
What emerged was not a gradual drift, but a structured process.
Two distinct waves of gene activity unfolded in sequence, reshaping the cell’s identity. The first wave rapidly altered chromatin, changing which parts of the genome were accessible and effectively resetting the cell’s regulatory landscape. The second wave then consolidated a broader shift in cell identity, moving cells away from a differentiated, drug-sensitive melanoma state toward a more primitive, therapy-tolerant condition.
“This is not a random process,” said Dr. Wei Wei, associate professor at ISB and co-senior author of the study. “The cells are executing a program. They are actively reprogramming themselves to survive.”

An Escape Route Built on Stress
At the center of this transition is a familiar biological signal: stress.
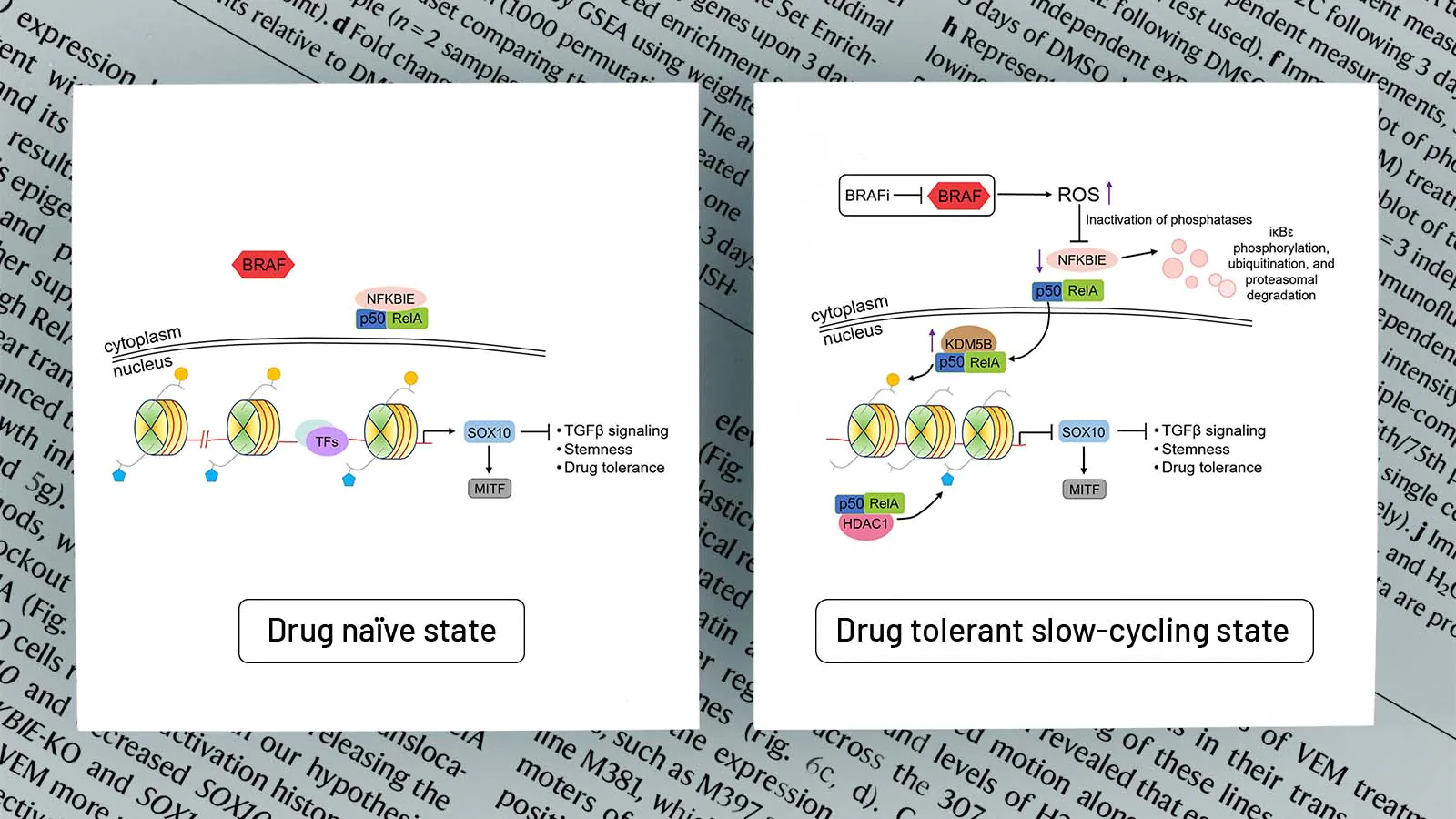
When melanoma cells are treated with BRAF-targeted therapies, the drugs do more than shut down a key growth pathway. They also disrupt the cell’s internal balance, leading to a buildup of reactive oxygen species — chemically reactive molecules that signal oxidative stress.
That stress activates NF-κB, a master regulator of cellular survival pathways.
NF-κB is not new to biology. First identified in the 1980s by Nobel laureate David Baltimore and colleagues, it has long been known as a central player in inflammation and immune signaling. What this study shows is that the same pathway first brought to prominence in foundational immunology reappears decades later as a key regulator of cancer’s adaptive response to treatment pressure.
Once activated, NF-κB moves into the nucleus and recruits epigenetic machinery — including enzymes that modify chromatin — to shut down genes that define the cell’s original identity. One of the key targets is SOX10, a transcription factor that helps maintain the melanocytic state.
As those identity programs are suppressed, the cells shift into a more flexible, drug-tolerant state that allows them to endure treatment.
Not Just Reversible — But Shaped by History
One of the study’s most striking findings is what happens when the drug is removed.
The cells can return to a drug-sensitive state — but they don’t simply rewind the process. Instead, they take a different path back, reflecting on where they have been.
This phenomenon suggests that cancer cells carry a kind of molecular memory of their treatment history. In other words, the transition is not just reversible — it is history-dependent. The cells retain a memory of prior stress, and that memory appears to be written, at least in part, into the epigenetic state of the system.
“This tells us that reversibility does not mean the tumor resets to its original condition,” said ISB President and Professor Dr. Jim Heath, co-senior author of the study. “It means the cells can move back, but with history embedded in the system. The tumor has learned something.”

A Broader Principle in Cancer
Although the study focused on melanoma, the researchers found evidence that similar stress-driven pathways operate in other cancer types, including lung and colon cancer.
That raises the possibility that this is not a niche mechanism, but a more general strategy that cancer cells use to survive targeted therapies.
The work also suggests that some tumors may be more “prepared” for this transition than others. Cells with more permissive chromatin — a more open and flexible regulatory landscape — appear better able to shift into the drug-tolerant state. That raises the possibility of an “epigenetic gauge of plasticity”: a way to read out, before or early during treatment, which tumors are most likely to adapt.
Why This Changes How We Think About Treatment Resistance
The classic view of targeted therapy is straightforward: hit the oncogene, shrink the tumor, and then worry later about the resistant clone that eventually emerges.
This work argues that the story starts much earlier. The first therapeutic blow already reorganizes the system. It suppresses the driver, but it also creates oxidative and signaling stresses that help launch a survival program. Resistance, in that sense, is not just the endpoint of mutation and selection. It begins as a dynamic state transition
That insight opens a different therapeutic possibility. Rather than focusing only on the endpoint — fully resistant tumors — the study highlights a critical window at the very start of treatment, when cancer cells first begin to adapt.
Combining targeted therapies with drugs that block the stress response or the epigenetic changes that enable this transition — preventing oncogene inhibition from becoming the very trigger of the escape state it is meant to suppress.
“If we can prevent cells from entering this escape state in the first place, we may be able to make therapies last longer,” Dr. Hui-Yu Chuang said, a co-first author of this study.
Seeing Cancer As a Dynamic System
More broadly, the study reflects a shift in how researchers think about cancer.
Instead of viewing tumors as static collections of mutations, this work treats them as dynamic systems — capable of reorganizing themselves in response to stress.
By combining experimental biology with computational modeling, the ISB team was able to capture these dynamics in detail.
“It is not just a story about melanoma. It is a story about how therapy itself can reshape the disease it is trying to destroy, how a pathway with deep roots in modern biology can resurface in a new role, and how cancer cells can remember the treatments that nearly killed them.” Wei said.
This project was supported by grants from the National Cancer Institute (NCI), the Andy Hill CARE Fund, and other funding agencies.
The study, “Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer,” was published in Nature Communications.


