 isbscience.org/research/genomic-architecture-of-inflammatory-bowel-disease-in-5-families-with-multiple-affected-individuals/
isbscience.org/research/genomic-architecture-of-inflammatory-bowel-disease-in-5-families-with-multiple-affected-individuals/Jan. 13, 2016
3 Bullets:
- In medical genetics it is an unsolved question to what degree complex diseases (such as inflammatory bowel disease) are influenced by rare variants with potentially large effects in relation to the many common and weak-effect variants that have been identified in genome-wide association studies (GWAS).
- By analyzing whole-genome sequences of five families with a high burden of inflammatory bowel disease (IBD) we aimed to elucidate the genomic architecture of IBD.
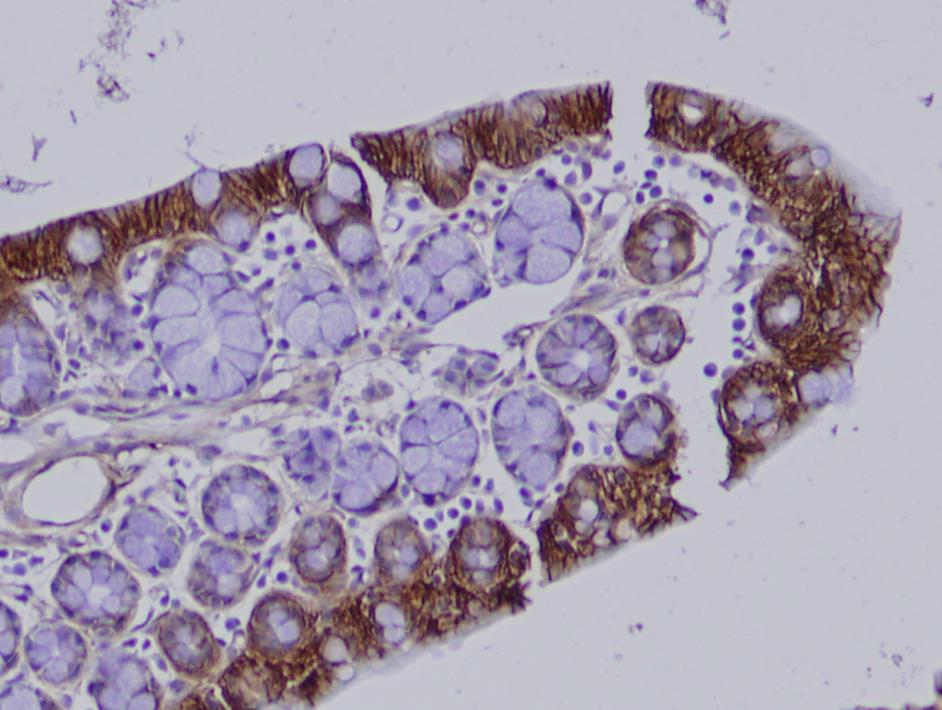
- We identified rare candidate variants and followed up one of them, a novel missense variant in TRIM11. We found evidence that the variant may foster IBD development by increasing inflammatory signaling in the gut.
By Anna-Barbara Stittrich
Our Family Genomics team at ISB together with gastroenterologists Steve Guthery, of University of Utah, and Richard Duerr, of University of Pittsburgh; and immunologist Jacques Peschon from the former Novo Nordisk Inflammation Research Center in Seattle; and other collaborators recently published a study on the genomic architecture of inflammatory bowel disease (IBD).
IBD is a complex, multifactorial disease characterized by chronic inflammation in the gastrointestinal tract. Since family history is the strongest risk factor for IBD a genetic contribution is apparent. Genome-wide association studies (GWAS) have been applied to many complex diseases in the last 10 years and revealed a plethora of disease-variant associations. For IBD, 163 variants have been identified in GWAS. However, immanent to the GWAS approach, these variants have only weak effects and are unsuitable to predict disease in an individual. It has been speculated that rare variants with high penetrance could contribute to IBD and other complex diseases. In this study we applied a family genomics approach and analyzed the full genomes of 38 individuals from five families, under the hypothesis that families with multiple IBD-affected individuals harbor one or more risk variants that (i) are shared among affected family members, (ii) are rare, and (iii) have substantial effect on disease development.
Our analysis revealed not only novel rare candidate risk variants, but also a high burden of common known IBD-associated variants in four out of the five families. In fact, the majority of the expected genetic contribution can be explained with known GWAS variants. However, functional analysis of our top candidate variant in the remaining family, a novel missense mutation in the ubiquitin ligase TRIM11, suggests that this variant leads to increased NF-κB signaling, a pathway that has been well implicated with IBD development before.
We conclude that high polygenic risk scores based on common weak-effect variants account for the high incidence of IBD in most, but not all families we analyzed, and that a family study design can identify novel rare variants conferring risk for IBD with potentially large effect size, such as the TRIM11 mutation.
Title: Genomic architecture of inflammatory bowel disease in five families with multiple affected individuals.
Publication: Human Genome Variation
Authors: Anna B Stittrich, Justin Ashworth, Mude Shi, Max Robinson, Denise Mauldin, Mary E Brunkow, Shameek Biswas, Jin-Man Kim, Ki-Sun Kwon, Jae U Jung, David Galas, Kyle Serikawa, Richard H Duerr, Stephen L Guthery, Jacques Peschon, Leroy Hood, Jared C Roach, Gustavo Glusman
Link: nature.com/articles/hgv201560